Cite CaviDB
CaviDB: a database of proteins cavities features for structure-based drug design
Ana Julia Velez Rueda, Franco Leonardo Bulgarelli, Nicolás Palopoli, Gustavo Parisi
CaviDB Overview
The first step to running CaviDB is providing a valid PDB ID, UniProt ID, UniProt Entry name or Alphafold ID and the web server will automatically load all the chains related to the search and their general data including their length, number of cavities predicted, and relevant cross-references identifiers.

The features obtained for each entry are organized in two main sections describing the general cavities descriptors, including an interactive display for cavities visualization, the inter-cavities contacts, and the activated residues per cavity, and the global protein descriptors.
Cavities Descriptors
All the cavities predicted for each entry can be visualized interactively, by selecting them in the cavities buttons. They will be highlighted in colors both, in the structure, and also in the sequence.
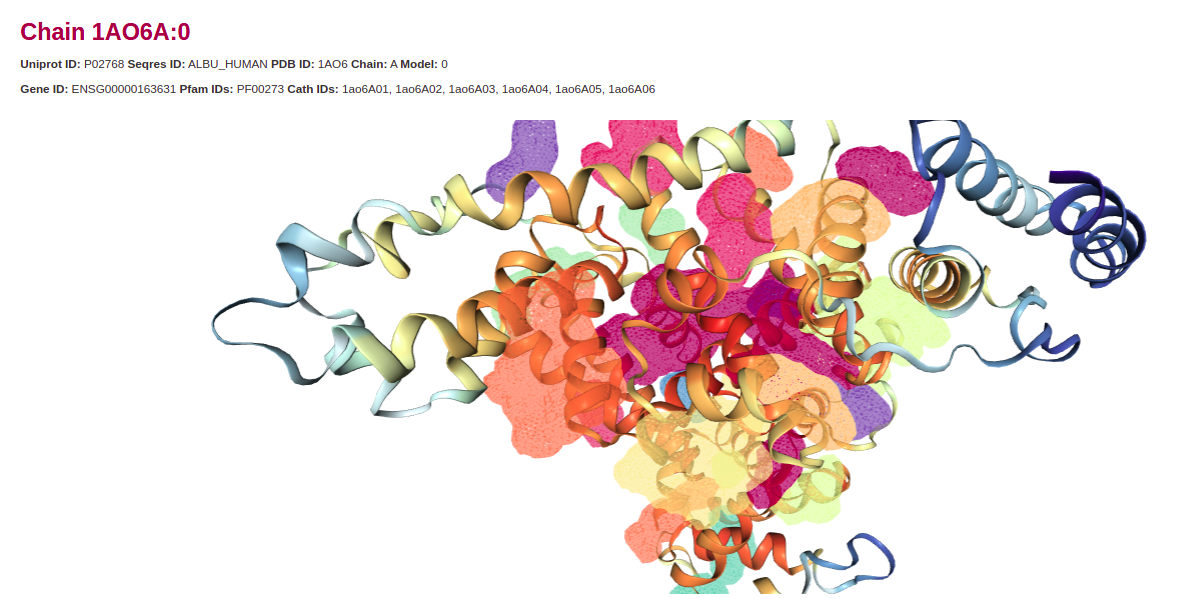
Druggable cavities (with a Druggability score > 0.5) are tagged with an icon , and can be selected or unselected with a button located before the sequence visualization.
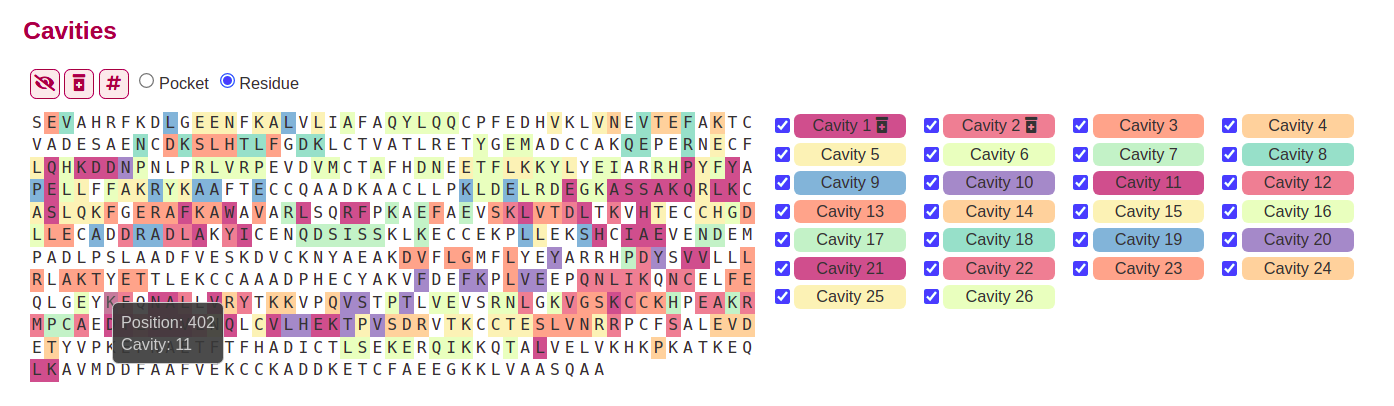
The primary structure of each entry can be displayed in two different representations: a letter soup, and periodic table-like visualization.
When selecting the # button, each residue is represented in boxes containing information about the residue type and its position in its center, the cavities in which it is part in the top left margin, and the number of contacts it has in the top right margin.
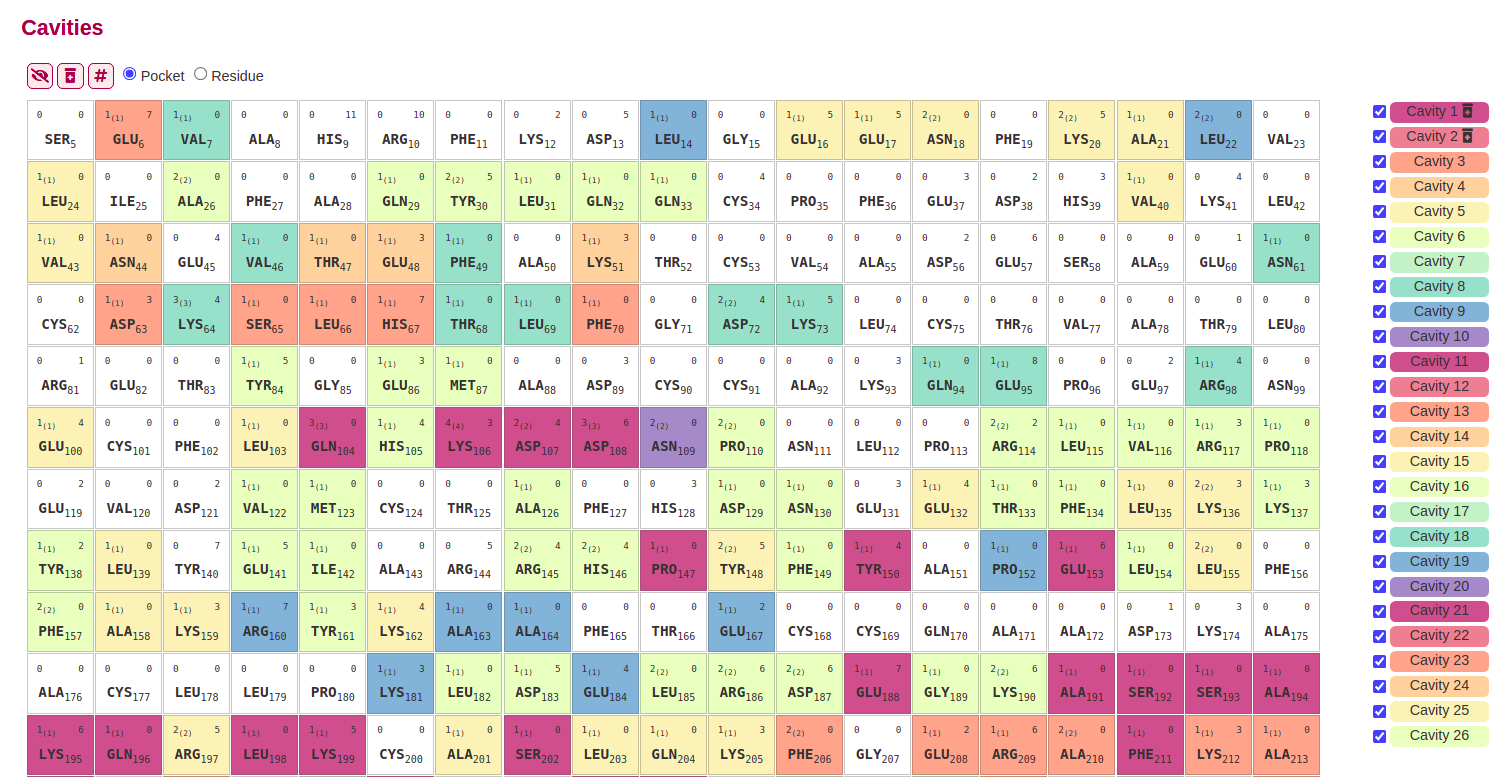
By default the # button is unselected, so the sequence is shown as a letter soup, in which the residues shows the information about its position and cavities when making hover in them.
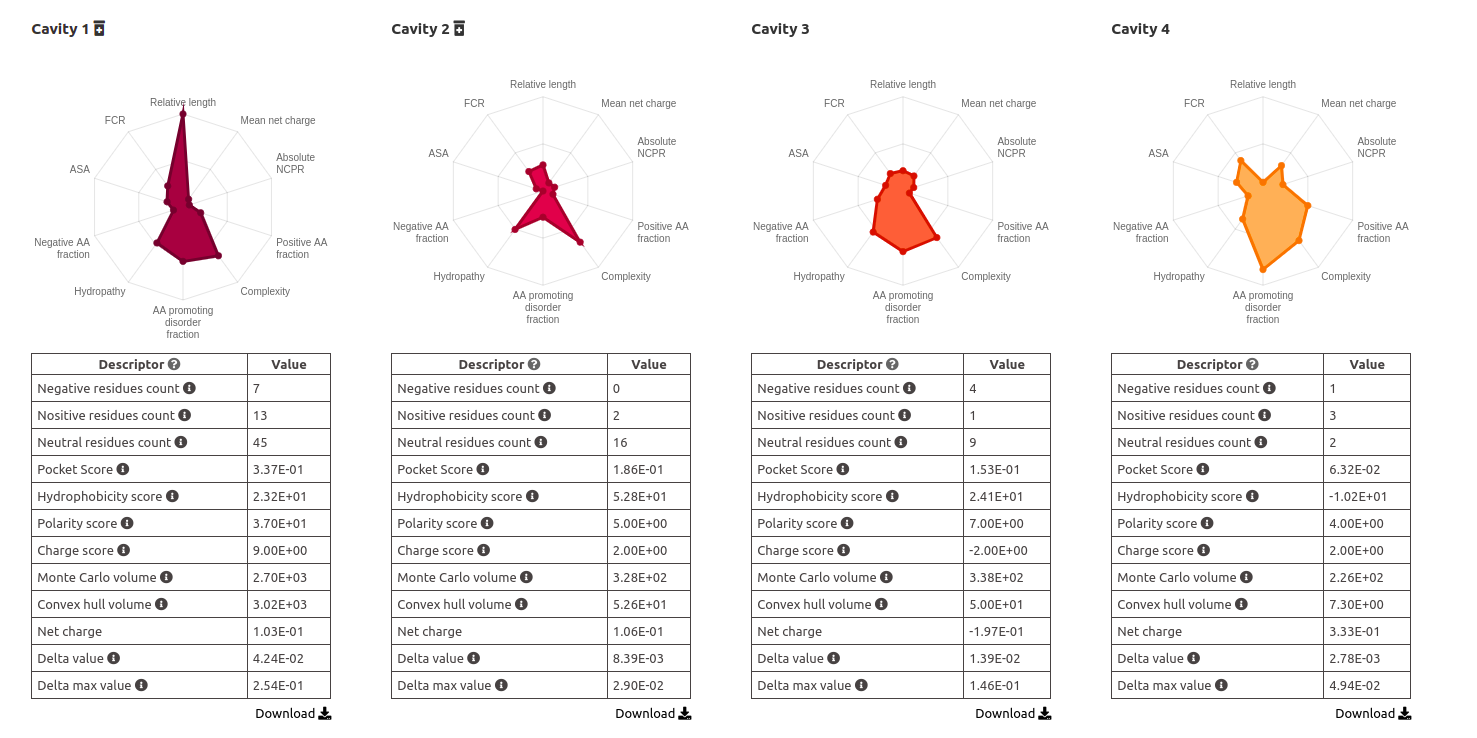
All the descriptors calculated by ,Fpocket are shown in tables and star plots, for their easy comparission.
Intercavities contacts
Intercavities contacts heatmap and network plot, with the druggable cavities highlighted with a black border are provided to users.
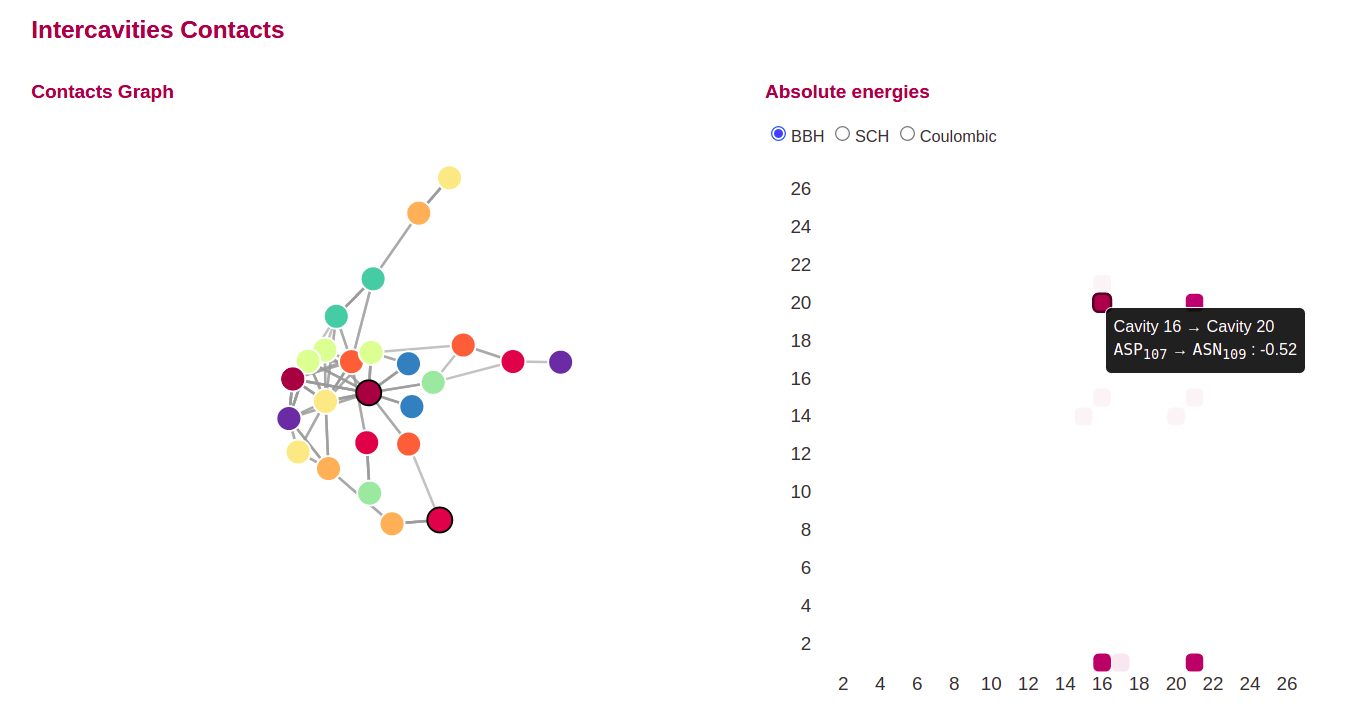
The residues linking the cavities and the binding energies corresponding to each contact is shown when making hover in the contacts matrices. All the contacts types predicted by Propka are shown in different contacts matrices: Side chain hydrogen bonds (SCHBOND), Backbone hydrogen bonds (BBHBOND), and Coulombic bounds.
Activated cavities
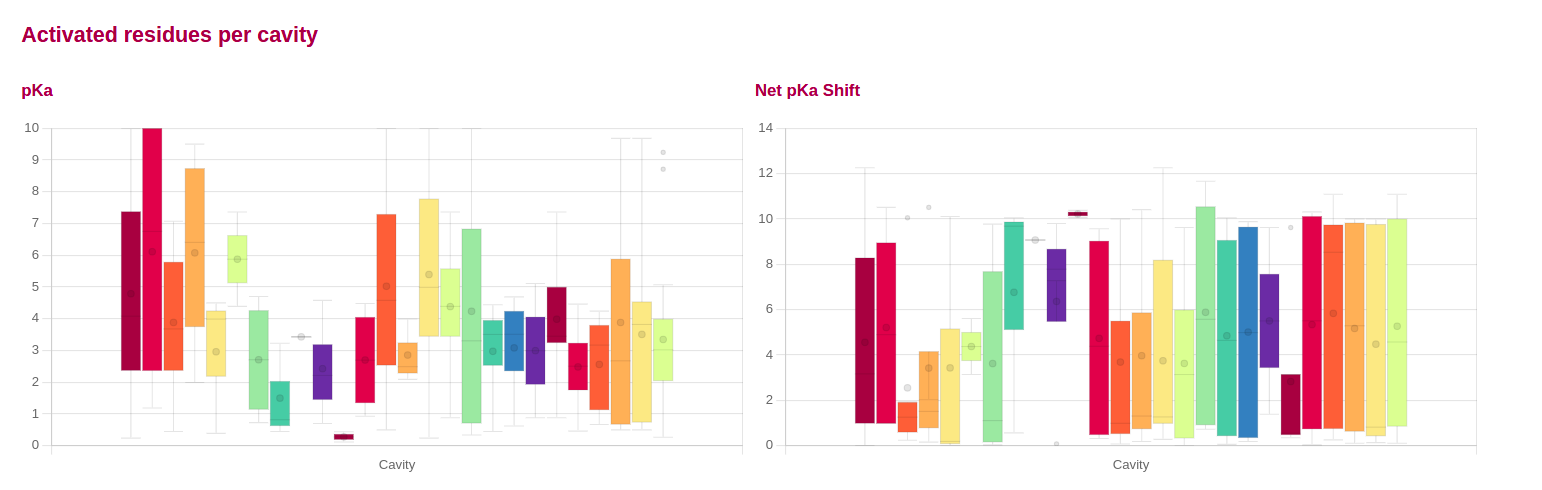
Information about the activated cavities and Net pKa shift per cavity, are displayed in different plots, colored by cavities colors.
CaviDB Advanced Search
There are many different options for searching CaviDB Entries and pre-filtering the results. It is possible to specify an Uniprot ID using the tag 'uniprot:'
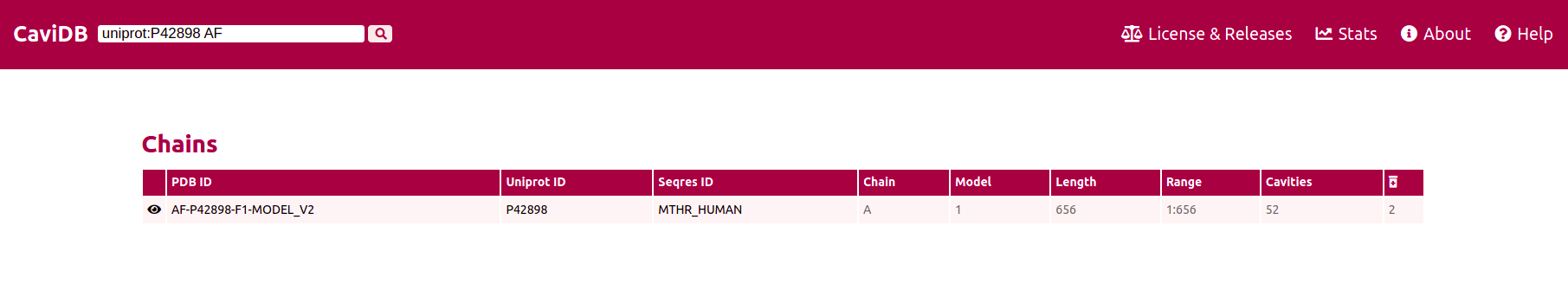
When combining the tag 'uniprot:' with the suffix 'AF', then only the Alphafold conformers of the required protein will be shown. By using the tag 'conf' and providing CaviDB with two specific PDB or Alphafold ids separated by a |, can be retrieved these two specific entries.
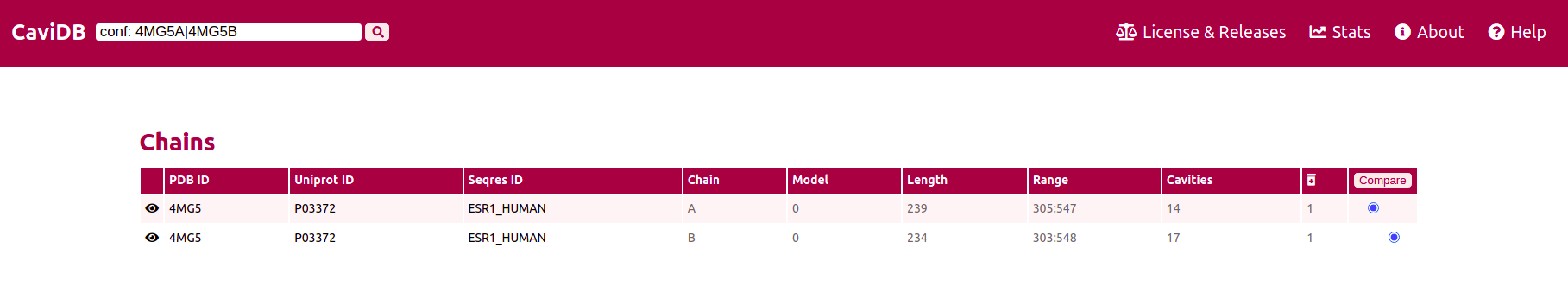
The two conformers can be selected for the comparison, and by clicking on the 'compare' button the difference between both will be renderized. The left plots and cavities selectors corresponds to the first conformer and the right to the second one.
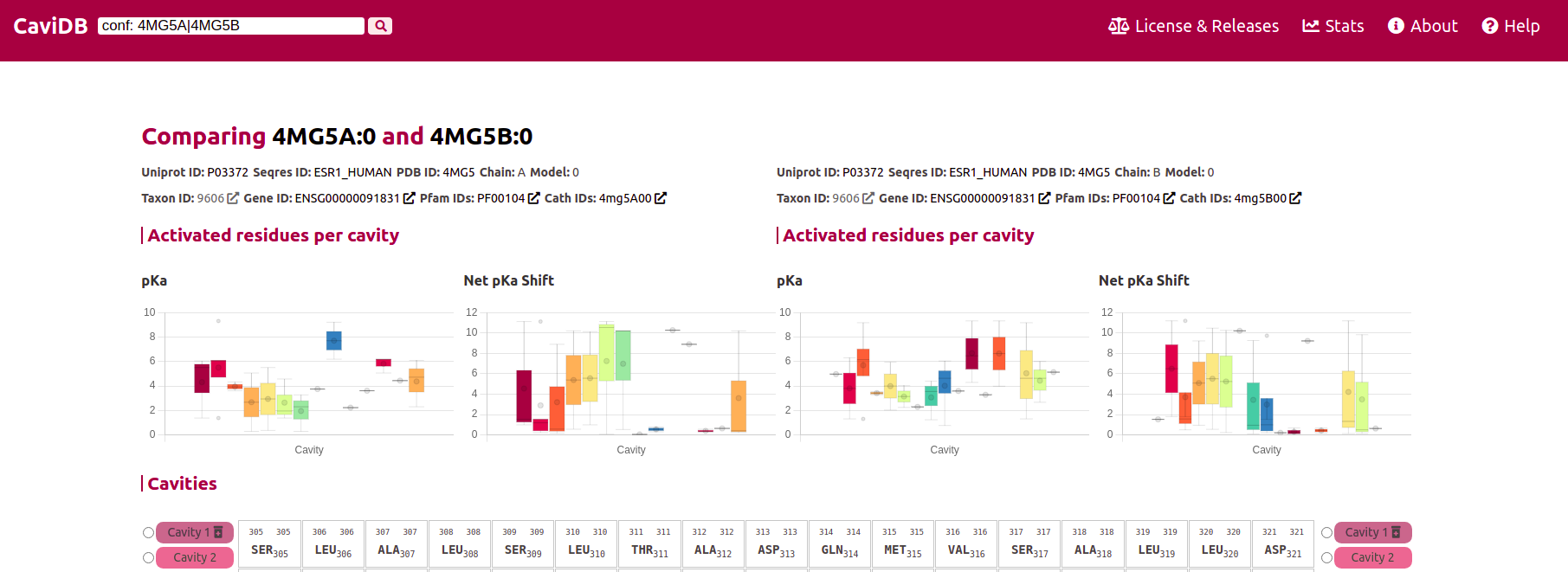
By clicking on the cavities selector from each conformer, the cavities will be colored in the sequence. Find more search examples in the following table:
| Search tag example | Result expected |
|---|---|
| pdb:1AO6 chain:A model:0 | Entry corresponding to 1AO6, chain A, and model 0 |
| 1AO6A:0 | Entry corresponding to 1AO6, chain A, and model 0 (shortcut) |
| ~1AO6A:0 | Entries corresponding to PDB 1AO6, chain A, and model 0 or an Alphafold model for the same Uniprot ID |
| 4MG5A|4MG5B | Entries corresponding to PDB ID 4MG5 chain A or 4MG5 chain B |
| uniprot:P51825 | Entries corresponding to UniProt ID P51825 |
| P51825 | Entries corresponding to UniProt ID P51825 (shortcut) |
| entry:Q4CPZ0_TRYCC | Entries corresponding to Q4CPZ0_TRYCC UniProt Entry Name |
| uniprot:Q4CPZ0 af | Alphafold entries corresponding to UniProt ID Q4CPZ0 |
| Q4CPZ0 af | Alphafold entries corresponding to UniProt ID Q4CPZ0 (shortcut) |
Descriptors detail
Cavities descriptors
| Descriptor | Value |
|---|---|
| Negative Residues Count | [CIDER] Get the number of negatively charged residues in the sequence (D/E) |
| Positive Residues Count | [CIDER] Get the number of positively charged residues in the sequence (R/K) |
| Neutral Residues Count | [CIDER] Get the number of neutral amino acids |
| Pocket Score | [Fpocket] Internal parameter |
| Hydrophobicity score | [Fpocket] Based on a residue based hydrophobicity scale published by Monera & al. |
| Polarity score | [Fpocket] Hydrophilicity character of a binding pocket |
| Charge score | [Fpocket] Charge of each amino acid in the binding site is tracked |
| Monte Carlo volume | [Fpocket] Feature based on Monte-Carlo algorithm calculations, full volume occupied by all alpha sphere in a given pocket |
| Convex hull volume | [Fpocket] This data resumes relative volume of different amino acids |
| Net charge | [CIDER] Get the net charge per residue of the sequence NCPR |
| Delta value | [CIDER] Returns the delta value of the sequence, as defined when calculating kapp |
| Delta Max Value | [CIDER] Returns the maximum possible delta value (delta-max) for a sequence of this composition |
For more information check the CIDER and Fpocket documentation
Protein global descriptors
| Descriptor | Value |
|---|---|
| Length | Sum of sequence characters |
| Count Neutral Res | [CIDER] Get the number of neutral amino acids |
| Delta value | [CIDER] Returns the delta value of the sequence, as defined when calculating kapp |
| Delta Max Value | [CIDER] Returns the maximum possible delta value (delta-max) for a sequence of this composition |
| Mean net charge | [CIDER] Get the absolute mean net charge of the sequence (pH=7) |
| Fraction of aminoacids expanding residues | [CIDER] Get the fraction of residues which are predicted to contribute to chain expansion (E/D/R/K/P) |
| Fraction of aminoacids promoting disorder residues | [CIDER] Get the fraction of residues predicted to be ‘disorder promoting’ |
| Aliphatic index | [MODLAMP] Method to calculate the aliphatic index of every sequence in the attribute sequences. |
| Aromaticity | [MODLAMP] Method to calculate the aromaticity of every sequence in the attribute sequences. |
| Predicted charge | [MODLAMP] Method to overall charge of the sequence (based on Bjellqvist method) |
| Charge density | [MODLAMP] Method to calculate the charge density (charge / MW) of the sequences |
| Hydrophobic ratio | [MODLAMP] Relative frequency of the amino acids A,C,F,I,L,M & V |
| Instability index | [MODLAMP] The instability index is a prediction of protein stability based on the amino acid composition ([1] K. Guruprasad, B. V Reddy, M. W. Pandit, Protein Eng. 1990, 4, 155–161.) |
| Isoelectric point | [MODLAMP] The isoelectric point of the sequence, based on the pK scale is extracted from CRC Handbook of Chemistry and Physics, 96th ed |
| Selectivity Index | [MODLAMP] Sequence selectivity index scale for helical antimicrobial |
| Argos hydrophobicity scale | [MODLAMP] Sequence Argos hydrophobicity |
| Bulkiness | [MODLAMP] Sequence bulkiness, based on amino acid side chain bulkiness scale |
| Eisenberg hydrophobicity scale | [MODLAMP] Sequence hydrophobicity based on Eisenberg consensus |
| Grantham side-chain descriptor | [MODLAMP] Sequence side chain composition, polarity and molecular volume |
| pepArc | [MODLAMP] Pharmacophoric feature scale, dimensions are: hydrophobicity, polarity, positive charge, negative charge, proline |
| T-scale | [MODLAMP] A PCA derived scale based on amino acid side chain properties calculated with 6 different probes of the GRID program |
| Extended five dimensional scale | [MODLAMP] The extended five dimensional Z-scale (z5) |
| Original 3D scale | [MODLAMP] The original three dimensional Z-scale (z3) |
| Intercavities Contacts | [Propka] BBH, SCH, and Coulombic binding energies between residues |
| Ionizable residues pKa | [Propka] Ionizable residues pKa |
For more information check the CIDER, Modlamp, and Propka documentation
Downloads
CaviDB provides Fpocket outputs for each cavity detected per entry, also the global protein features can be downloaded.